SERVICES
- Bioinformatics analysis of high-throughput biological data, including proteomics, using our well-defined analysis pipelines
- Consulting services for custom analysis beyond our standard pipelines, including genome-wide association studies
- Grant writing assistance, including assistance fully developing the bioinformatics components of a grant, cost analysis, letters of support, and documenting the availability of tools and expertise to complete the research indicated
- Free training sessions in bioinformatics topics: visit the CRI Seminar Series to find out about upcoming classes and download materials from past sessions
METHODOLOGY
The Bioinformatics Core’s work combines the use of powerful computing resources, advanced analytics tools, and a commitment to security to transform large amounts of raw data into meaningful results. We use the CRI’s high-performance computing cluster and large-scale storage resources, with which complex analytics can be executed in parallel or distributed environments to produce fast data processing rates, improving application performance and cost effectiveness. Through advanced high-throughput analytics solutions, we dig down to the root of each computational challenge and design the most direct path to a solution. All our work is protected with the CRI’s automated, resilient backup and security systems, ensuring data integrity and access controls that are aligned with standards required by HIPAA, FISMA, and other regulations.
LET’S GET STARTED
Planning an experiment? The Bioinformatics Core has created a guide to Bioinformatics Experimental Design to help you get the best results.
Upon receiving a project request, the Bioinformatics Core will draft a proposal for the researcher that includes the scope of deliverables, a timeline for completion, and the estimated cost. The proposal will be formalized into a service level agreement (SLA), the SLA should be reviewed carefully to ensure it meets your needs before accepting. Once we receive the signed SLA, we will commence the project. The execution of the project is guided by frequent discussion between researchers and bioinformaticians, with progress updates provided regularly. Project results are delivered in the form of a written report. See here for our standard pipeline workflow and delivery policy.
Questions? Email us here.
Have you worked with us before? We’d love your feedback.
OFFICE HOURS
Interested in live one-on-one support with your bioinformatics research needs? Join us for our weekly office hours hosted every Tuesday by our Associate Director of Bioinformatics, Yan Li.
Every Tuesday
12:30pm – 3:30pm
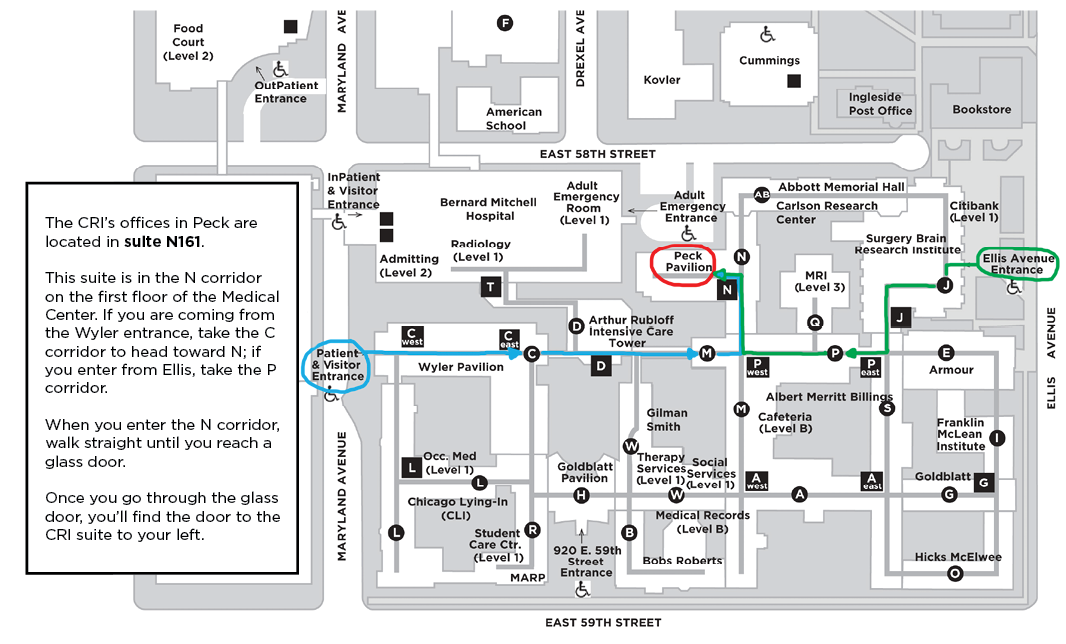
Medical Campus, Peck Pavilion Map
920 East 59th Street
Suite N161
Chicago, IL 60637
General Questions? Email us here.
Stay up to date by viewing CRI’s full event calendar.
ANALYSIS PIPELINES
We currently offer bioinformatics pipelines for genomic and proteomic data.
GENOMICS
Illumina
- RNA-Seq: Raw Data QC, Filtering, Mapping, Data Summarization, Expression Quantification, Differentially Expressed Genes, Pathways, and Gene Ontology Analysis, the analysis workflow, timeline and results delivery policy can be seen here.
- ChIP-Seq: Raw Data QC, Filtering, Mapping, Peak Calling, Peak Differential Analysis, Peak Related Genes Analysis, Gene Ontology Analysis, and Annotation
- Exome Sequencing: Raw Data QC, Pre-processing, Mapping with 3 different tools, Realignment and Quality Recalibration, Multiple Samples Variant Calling, Variant Annotation, Varian Comparison, Filtration, and Summarization
- Whole Genome Re-Sequencing (WGRS): Raw Data QC, Filtering, Mapping, Genotyping, SNP Detection, InDel Detection, SV (Somatic SV) Detection, CNV Analysis, and Annotation
- Consensus Genotyping Pipeline: Genotyping, SNP Detection and InDel Detection using three different methods (Samtools, GATK and Atlas-2), comparison of variant calls, list of consensus call variants, and list of method specific calls
- De-novo Assembly: Raw data QC, Merging, Clipping, Filtering, Contigs Assemble, Scaffold Assembly, Assemble Statistics, and Downstream Analysis
- Somatic Mutation Detection for Tumor/Normal Pairs: Raw Data QC, Pre-processing, Mapping with 2 different tools, Realignment and Quality Recalibration, Somatic Mutation Detection with 4 different tools, Variant Annotation, and Summarization
- Small RNA-Seq: Raw Data QC, Filtering, Mapping, Data Summarization, Quantification, Detection of Differentially Expressed miRNAs, Putative Gene Target Prediction, and Pathways Analysis
Illumina and Affymetrix Expression Arrays
- Filtering, Data Summarization and Normalization, Sample/Gene/Probe-based QC, Differentially Expressed Genes, Functional Annotation, and Pathway Enrichment Analysis
Affymetrix and Exiqon miRNA Arrays
- Filtering, Data Summarization and Normalization, Sample/Gene/Probe-based QC, Differentially Expressed miRNAs, Predict miRNA Targeted Genes, Functional Annotation, and Pathway Enrichment Analysis
scRNA-seq pipeline
- We provide bioinformatics analysis service for single-cell transcriptomic sequencing data generated by different technologies, including UMI-based Drop-seq and 10x Genomics Chromium, as well as non-UMI-based CEL-seq and Smart-seq. Our pipeline consists of (1) data de-multiplexing; (2) read alignment; (3) processing of cellular and molecular barcodes; (4) read count table; (5) quality control and normalization; (6) removal of debris, dead cells, and doublets; (7) detect batch effects; (8) clustering and identification of cell subsets; (9) cluster visualization using tSNE or UMAP; (10) marker gene identification; (11) differential gene expression analysis; (12) GO term and pathway analysis. Our customized pipeline can also perform single cell lineage analysis and other study-specific analyses upon request.
PROTEOMICS
Proteomics Analysis Pipeline
- Raw MS data processing, deisotoping, data conversion, spectral filtering, spectral library creation, extracted ion chromatogram generation (MASIC), peptide and protein identification (X!Tandem, MaxQuant, Mascot, MSGF+), quantification (MaxQuant, Scaffold), targeted assay generation (Skyline), and dataset alignment and feature generation (MultiAlign)
CyTOF pipeline
- We provide bioinformatics analysis service for single-cell Mass Cytometry (CyTOF) data. Our pipeline consists of (1) pre-gating (removal of debris, dead cells, and doublets); (2) diagnostic analysis; (3) marker ranking; (4) clustering, identification, and annotation of cell subsets; (5) cluster visualization using tSNE or UMAP; (10) differential analysis, including differential cell population abundance, differential analysis of marker expression stratified by cell population, and differential analysis of the overall marker expression. Our customized pipeline can also be incorporated into other study-specific analyses upon request.
Additional Services
- Profiling of post-translational modifications (phosphorylation, glycosylation, etc.), statistical analysis for labeled and label-free proteomics, and pathway enrichment analysis (Ingenuity Pathway Analysis)
We also offer custom-made pipelines and expertise for types of analysis not listed above, including genome-wide association studies. Email us to get started!
TOOLS AND DATASETS
The Bioinformatics Core maintains the following catalogs of software tools, reference datasets, and databases for use by the BSD research community:
MEET THE BIOINFORMATICS CORE
Our team of bioinformaticians offer a diversity of experience and areas of expertise.
 Mengjie Chen, PhD
Mengjie Chen, PhD
Faculty Director, Bioinformatics core
mengjiechen@uchicago.edu
Mengjie Chen is an Associate Professor of Genetic Medicine, Human Genetics and Statistics at the University of Chicago. Before joining UChicago, she was an assistant professor in the Department of Biostatistics and Genetics at UNC-Chapel Hill from 2014 to 2016. She obtained her PhD in Computational Biology and Bioinformatics from Yale University in 2014. Dr. Chen was a recipient of the Alfred P. Sloan Research fellowship in Computational and Molecular Evolutionary Biology in 2019. As a computational biologist and statistician by training, Dr. Chen’s research bridges statistical methodological advances and biomedical applications. Her group develops computational methods and open source tools to address challenges posed by high-throughput technologies for data analysis and interpretation. She has led analysis for numerous genomics projects, including single cell RNA-seq analysis for female reproductive systems in Human Cell Atlas project. Other scientific highlights in cancer genomics include: profiling esophageal squamous-cell carcinoma for Asian population for the first time and profiling matched primaries and multiple metastases from 16 breast cancer patients. As Faculty Director, Dr. Chen oversees development of all bioinformatics projects, facilitates on-campus team science, and sets overall policy/infrastructure for the Bioinformatics Core Facility.
 Wenjun Kang, MS
Wenjun Kang, MS
Technical Director, Senior Bioinformatician
wkang2@bsd.uchicago.edu
Wenjun joined the CRI Bioinformatics Core in 2012. He has extensive experience in developing web-based applications for clinical studies, molecular pathology labs, and project management. His areas of expertise also include biostatistics analysis, study design, and software development. Wenjun’s preferred languages are Python/Django, jQuery, MySQL for web development, and SAS/R for statistical analysis. He holds master’s degrees in Biostatistics and Health Informatics from the University of Minnesota.
 Yan Li, PhD
Yan Li, PhD
Associate Director of Bioinformatics
yli22@bsd.uchicago.edu

Yan received her PhD in Bioinformatics (December 2013) and dual MS degrees in Statistics and Plant Pathology (August 2009) from the University of Georgia. Prior to joining the CRI in 2014, she worked on multidisciplinary projects using advanced computational tools, statistical methods, and mathematical modeling to address a variety of questions across the biological sciences. Currently, she is focused on developing and applying bioinformatics computational software and pipelines to facilitate the analysis of Next Generation Sequencing (NGS) data. She works closely with faculty and researchers to explore genomics-associated biomedical questions, including the NGS data analysis of RNA-Seq, ChIP-Seq, small RNA-Seq, and exome sequencing.
 Houxiang Zhu, PhD
Houxiang Zhu, PhD
Bioinformatician
houxiang@bsd.uchicago.edu

Houxiang received his PhD in Bioinformatics (2019) from Miami University. Prior to join the CRI Bioinformatics Core in 2022, he worked as a postdoctoral research associate at Washington University in St. Louis. Houxiang has a lot of experience in web application development and Next Generation Sequencing (NGS) data analysis, including WGS, WES, RNA-seq, and scRNA-seq. Currently, Houxiang mainly focuses on NGS data analysis and pipeline development.
 Evan Wu
Evan Wu
Bioinformatician
evanwu@uchicago.edu

Evan obtained his bachelor’s degree in biological sciences from the University of Chicago. His current work revolves around RNA-seq analysis and pipeline development, as well as the development of web-based applications for bioinformatic and clinical usages. He additionally has expertise in statistical genetics: using machine learning and deep learning methods to understand the role of human genetic variation in complex traits and diseases.
 Jason Shapiro, PhD
Jason Shapiro, PhD
Bioinformatician
shapiro@bsd.uchicago.edu
Jason received his PhD in Ecology and Evolutionary Biology from Yale University (2014). Prior to joining the CRI Bioinformatics Core in 2021, he was a postdoctoral research associate at Loyola University Chicago, where he developed computational methods to explore the diversity and evolution of bacteriophages. His research currently focuses on virus comparative genomics. Jason has experience working with a range of data types and bioinformatic questions, including microbiome analysis, genome assembly, network analysis, variant analysis, and transcriptomics, including single-cell and spatial datasets.

Diana Vera Cruz, PhD
Bioinformatician
dveracruz@bsd.uchicago.edu
Diana received her Ph.D. in Computational Biology and Bioinformatics (May 2020) from Duke University. Before joining the CRI Bioinformatics core in 2022, she was a research scientist in the Department of Ecology and Evolution at the University of Chicago. Diana studied viral evolution across scales and how it is impacted by immune selection, antibody dynamics, and vaccination. Her expertise includes population genetics, phylogenetics, mathematical modeling, statistical methods, NGS data analysis, and pipeline development.
 Qiaoshan Lin, PhD
Qiaoshan Lin, PhD
Bioinformatician
Qiaoshan.Lin@bsd.uchicago.edu

Qiaoshan received her Ph. D. in Ecology and Evolutionary Biology from the University of Connecticut. Prior to joining the CRI Bioinformatics Core in 2022, she studied developmental genetics in Monkeyflowers combining wet and dry approaches. Her bioinformatic experience includes de novo genome assembly, genome annotation, comparative genomics, variant analysis, and workflow development. She is also skilled in RNAseq and small RNAseq data analysis.

Yildiz Koca, PhD
Bioinformatician
yildizkoca@bsd.uchicago.edu
Yildiz got her Ph.D. in Biomedical Sciences in 2020 from Icahn School of Medicine at Mount Sinai. Before joining the CRI Bioinformatics Core in 2022, she had been a postdoctoral associate at Yale School of Medicine. Yildiz studied how signaling pathways regulate cell specification and morphogenesis during development in different model organisms. Her expertise includes developmental biology, genetics, cell biology and NGS data analysis.

Zhongyu Li, MS
Bioinformatician
zhongyu1@bsd.uchicago.edu
Zhongyu received her Master’s degree in Biomedical Engineering from Rice University. She is currently doing her second master’s in Computer Science at Georgia Tech. Before joining CRI, she worked as a Translational Clinical Research Specialist at Houston Methodist Hospital, focusing on medical imaging and robotics. She worked on multiple industry-collaborated pre-clinical projects. She is passionate about clinical algorithm optimization and web-based application development.

David Tieri, PhD
Bioinformatician
David.Tieri@bsd.uchicago.edu
David received his PhD in Physics from the University of Colorado. Before joining CRI, he worked as a senior research associate in the department of Biochemistry and Molecular Genetics at the University of Louisville. There, he used single cell technologies and adaptive immune receptor repertoire sequencing for immune profiling studies in the context of disease. He also studied how the genetics of Immunoglobulin locus affects the adaptive immune receptor repertoire and hence immune response. He is currently interested in using deep learning models to make functional predictions such RNA sequence coverage from genetic sequence, and how these models can be applied in the context of immune response. He is passionate about partnering with clinicians to translate his scientific and data analysis skills into the clinic to aid in the development of the next generation of personalized immunotherapies.

Geetha Priyanka Yerradoddi, MS
Bioinformatician
Geetha.Priyanka@bsd.uchicago.edu
Geetha Priyanka received her Master’s degree in Bioinformatics from Georgia Institute of Technology in 2023. Prior to joining CRI Bioinformatics core, she worked as Graduate Research Assistant at Georgia Tech with experience in Genomics, population genetics, NGS Data analysis and statistical analysis. Additionally, she has corporate experience focused mainly on pipeline development, and R Shiny applications. Devoted to gaining expertise in scRNA seq, ATAC-seq, ChIP-Seq and many other cutting-edge sequencing advances. Passionate about learning and incorporating Machine learning and AI models that advance our understanding of human diseases.

{kind=link}
Katie Aracena, PhD
Bioinformatician
Katherine.Aracena@bsd.uchicago.edu
Katie received her Ph.D. in Human Genetics from the University of Chicago in 2023. Her research leverages genetic, functional genomics, and clinical datasets to understand molecular mechanisms underlying human disease. She has experience analyzing and integrating genetic, transcriptomic, and epigenomic data types including whole-genome sequencing (WGS), RNA-Seq, ATAC-Seq, ChIP-Seq and DNA methylation data.
Interns
- Michiko Ryu (2021) master student in Physical Science at U Chicago.
- Qing Gong (2021-2022) master student in Applied Statistics at Loyola University
- Xizhi Xu (2021-2022) undergraduate student in Statistics at University of Iowa.
- Dante Vairus (2022- ) high school student at U Chicago Laboratory Schools
- Tianhong Wang (2022-) undergraduate student in Data Science at University of California Irvine
Past Fellows
- Lei Huang, PhD, Senior Bioinformatician, 2013-2020, now a Senior Research Scientist at Kite Pharma
- Ziyou Ren, PhD, Bioinformatician, 2020-2022, now an Assistant Professor at Northwestern University
- Chang Chen, PhD, Bioinformatician, 2020-2022, now a Scientist at FDA